Our Research
We have a broad interest in investigating how environmental change drives evolution at the population and species level, and in combining genomic and ecological techniques to connect gene flow processes at the individual and population level to macroevolutionary patterns, with a focus on species of conservation concern.
In addition to these research themes, we are advocates for reproducible research and open science in ecology and evolutionary biology, and work to teach others about the importance of these principles.
Population connectivity and landscape genomics of lowland leopard frogs
In this project, we are using ddRAD data for lowland leopard frogs (Lithobates [Rana] yavapaiensis) sampled in southeastern Arizona to assess the population genetic structure and connectivity of these frogs. Understanding connectivity among lowland leopard frog populations will provide information to guide future translocation and conservation efforts. This project is in collaboration with Dr. Javan Bauder, the Arizona Game & Fish Department’s Ranid Frogs team, the Pima County Conservation Lands and Resources Department, Coronado National Forest, Saguaro National Park, and The Nature Conservancy.
Popular Media about this project: “Unlocking Genetic Secrets for Arizona’s Lowland Leopard Frogs” (June 2024)



Photos pictured were taken by Beth Hasl and Jessica Rick.
Population connectivity and hybridization in Arizona-native Catostomid fishes
We are conducting a genomic study to understand the relationships among populations of desert suckers (Catostomus clarkii), Sonora suckers (Catostomus insignis), and their hybrids, seeking to understand (1) genetic connectivity among populations of desert sucker, (2) genetic connectivity among populations of Sonora sucker, (3) the rate of hybridization among the two species in streams where they co-occur, and (4) spatial variation in the rates of hybridization among the two species, and how this variation relates to stream characteristics (including anthropogenic influences) and ecological outcomes of hybridization.
To answer these questions, we captured native suckers in a variety of streams across the state of Arizona, took photos (for phenotyping), small fin clips (for genetic data), fecal samples (for disease and microbiome data, from a subsample of adult fish), and release all captured fish back into their habitats. With these data, we generated ddRAD data, from which we will calculate relatedness and genetic distance between all individuals within each species, allowing us to estimate genetic diversity within each drainage sampled, estimate connectivity between different streams, and estimate distinctiveness among fish from different river drainages. These efforts to quantify spatial variation in genetic differentiation and diversity will be important information for understanding the native sucker populations in Arizona and informing any future management and translocation efforts.

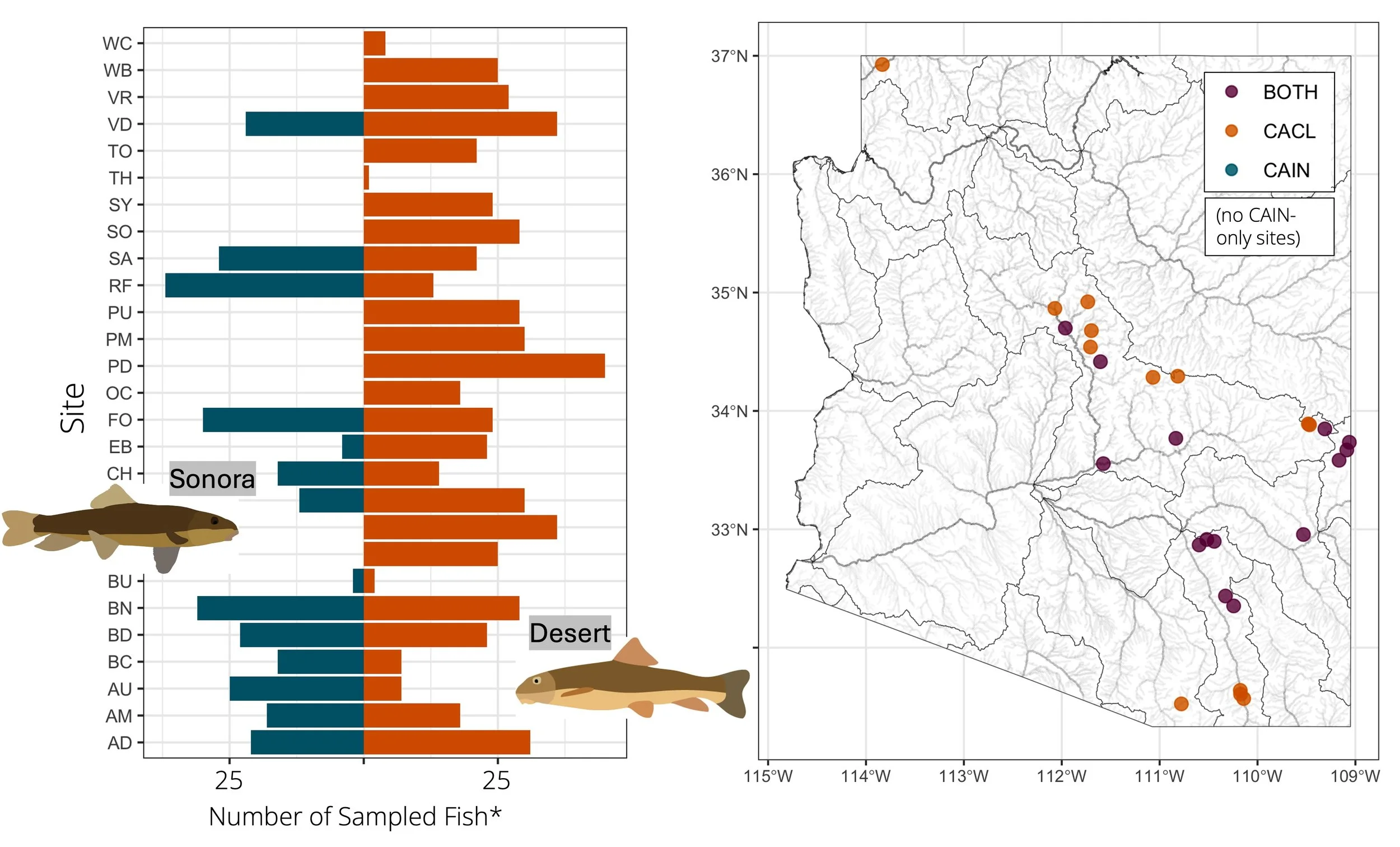
Sampling site locations and numbers of fish from the summer 2024 field season. Fish were identified in the field as either Sonora or desert suckers; however, these IDs may change once we analyze the genomic data.
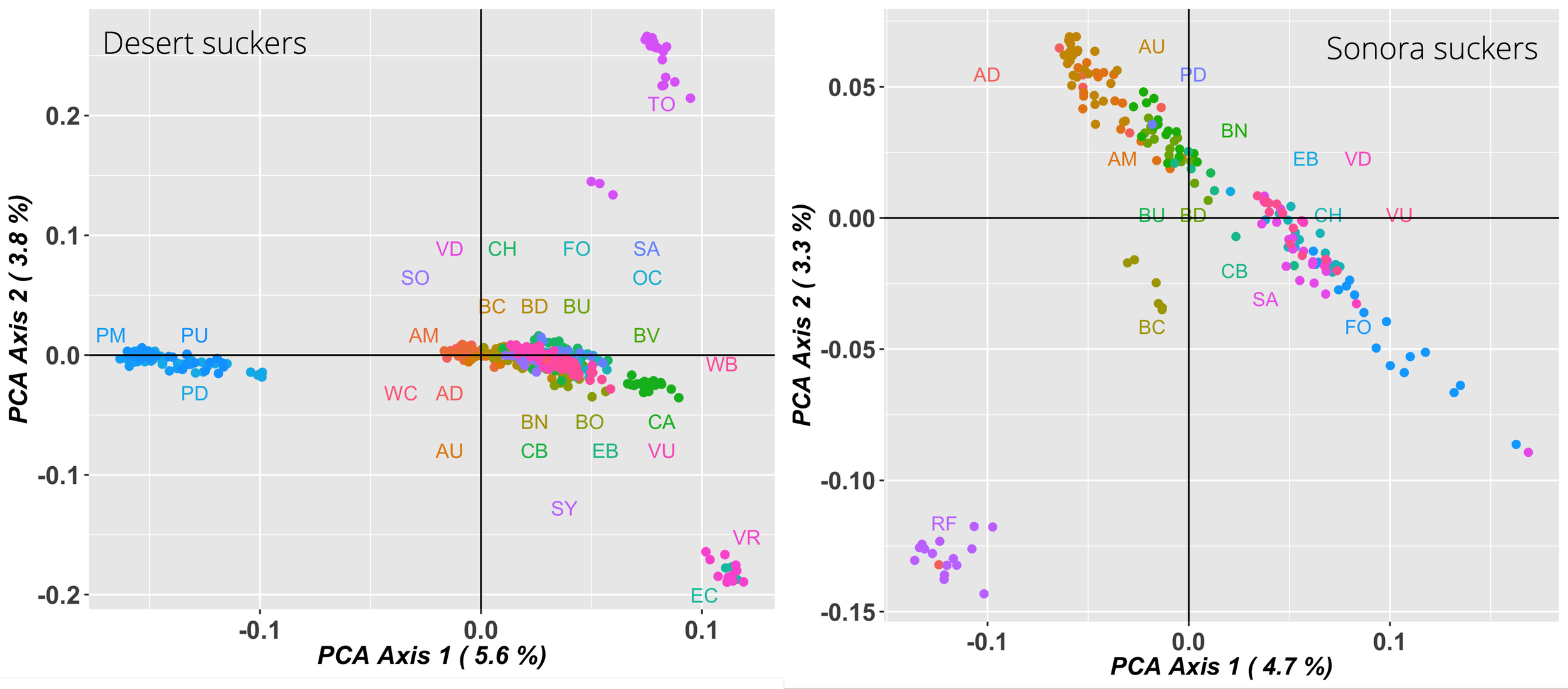
First-pass principal components analysis of the Desert sucker (left) and Sonora sucker (right) individuals that were sampled during the 2024 field season. Each point represents one individual fish, points are colored by the sampling location, and two-letter location codes are used to label each sampling location. Dots that are close to one another on the plot are individual fish that are closely related to one another genetically, while dots farther apart are fish that are less similar genetically.
Lesser long-nosed bat Kinship analysis
Lesser long-nosed bats (Leptonycteris yerbabenuae) have been observed to feed at hummingbird feeders in the Tucson area. In collaboration with Dr. Ted Fleming (professor emiritus, University of Miami), we are using genomic data to identify relationships among bats that were netted at hummingbird feeders in Tucson in 2023 and 2024. These data will be paired with radio transmitter location data to better understand foraging dynamics in these bats of conservation concern.
ROLE OF INVERSIONS IN FACILITATING LOCAL ADAPTATION
As a continuation of Jessi’s NSF Postdoctoral Fellowship working with Nina Overgaard Therkildsen at Cornell University and Hannes Baumann at University of Conneticuit-Avery Point, we are investigating the evolutionary mechanisms that facilitate local adaptation via chromosomal inversions in the Atlantic silverside study system, using a combination of common garden experiments, genomic investigations, and simulation studies.
Natural environments are often heterogeneous, with strong spatial variation in conditions within a species’ range. Populations within a species gain an advantage by adapting to these differing environments, and such local adaptation can be facilitated by structural variation within an organism’s genome. We are investigating how structural genomic variation allows populations to adapt to their environments, as well as how these types of variation enable population divergence and speciation. This research is important to understanding the mechanisms involved in the origin and maintenance of biodiversity, and how species may adapt to changing climatic conditions.
This research integrates data from common garden experiments, genomic analyses, and evolutionary modeling to investigate how structural genomic variation—specifically, inversions—facilitates local adaptation in wild populations. We are making use of the large inversion regions associated with locally adapted phenotypes in the Atlantic silverside (Menidia menidia) as a model system. Key outcomes of this project include: (1) identifying regions within inversions associated with local adaptation phenotypes using a common garden framework; (2) a population-wide selection scan to determine whether differentiation is greatest at inversion breakpoints or at candidate loci within inversions; and (3) simulations to quantify the conditions within which recombination suppression within inversions is a viable evolutionary mechanism for maintaining local adaptation. To achieve these outcomes, Atlantic silversides will be collected and raised in a common garden experiment, and phenotypic and whole genome data from these individuals will be collected for analyses.












Photos taken by Jessi Rick, as well as collaborators Hannes Baumann, Maria Akopyan, and Nina Therkildsen.
Phylogenetics and population history of Lates fishes

Lates stappersii

Fishing boat near Kigoma, Tanzania

Lates mariae
In collaboration with Katie Wagner and the Tanzanian Fisheries Research Institute in Kigoma, Tanzania, we are investigating the evolutionary history of endemic Lates (Nile perch) species in Lake Tanganyika, including divergence times, phylogenetic relationships, and admixture histories, as a method of contextualizing current declines in these fisheries-important fishes. In this research, we are combining population structure, demographic modeling, admixture, and phylogenetic analyses, and are combining various types of genome-scale data for non-model species (e.g., genotyping-by-sequencing, low-coverage whole genome sequencing, and reference-quality genome sequencing).
Related publication: Rick, JA*, J Junker*, PB McIntyre, I Kimirei, EA Sweke, JB Mosille, C Dinkel, S Mwaiko, O Seehausen, CE Wagner. The genetic population structure of Lake Tanganyika’s Lates species flock, an endemic radiation of pelagic top predators. Journal of Heredity, doi:10.1093/jhered/esab072. *equal contribution — also available as a preprint at https://doi.org/10.1101/2021.04.23.441176
Reference bias in SNP-based phylogenetics
Analysis of genomic data requires researchers to make a priori decisions on which data to retain and which to filter out, which can introduce biases into the data used in analyses. It is important to understand biases introduced by these data processing steps, which can substantially affect subsequent analyses. There has been little investigation into biases introduced by the choice of which reference genome(s) to use for aligning reads when using reduced-representation or whole genome resequencing data, yet these early steps may substantially alter inferences by systematically biasing which genetic loci are retained after processing. Together with Dr. Chad Brock, we are using simulations to investigate the effects of the distance from a reference genome to the in-group taxa (i.e., whether the reference genome is an in- or outgroup individual) and the manner in which this interacts with bioinformatic filtering choices made before analysis even begins in earnest.
Related publication: Rick, JA, CD Brock, AL Lewanski, J Golcher-Benavides, CE Wagner. 2024. Reference genome choice and filtering pipeline jointly influence SNP-based phylogenetic analyses. Systematic Biology, https://doi.org/10.1093/sysbio/syad065